
Matysiak, Silvina
EDUCATION
Ph.D., Rice University, 2007
Proteins are the nanomachines of biology. They possess a well-defined functional native conformation that is stable within a limited range of temperatures, pressures, and solvent conditions. This well-defined native structure is determined entirely by the protein's amino acid sequence, but a detailed understanding of how sequence determines structure remains elusive. Furthermore, several diseases including Alzheimer's and Parkinson's are associated with the formation of amyloid fibrils by self-assembly of misfolded proteins. Proteins are not static structures; in order to function, they have to move within their energy landscape, so a detailed characterization of the folding landscape is critical and would aid in the design of new proteins for a variety of applications, including pharmaceuticals, enzyme catalysis and biomaterials.
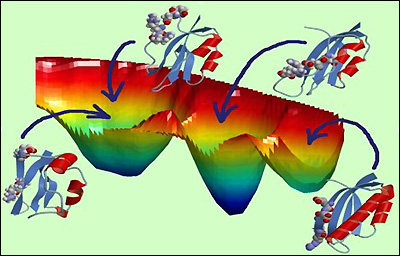
Free energy landscape of mutated protein S6 exhibiting the competition between protein folding and misfolding.
The study of biomolecular dynamics poses outstanding challenges both for theory and experiment. From the bio-chemical perspective, proteins, DNA, and RNA are giant molecules, comprised of thousands of atoms, and interacting with thousands of water molecules. Solvation/ desolvation strongly affect biomolecular folding transitions and almost all binding and docking events in biomolecular systems. Characteristic time-scales in protein systems span 16 orders of magnitude, from vibrations to highly cooperative structural transitions. Linking these vastly different time and length scales is a grand challenge in computational bioengineering. Our research activity is centered on the application of statistical mechanics techniques, molecular modeling and simulation to the study of biomolecular dynamics, folding, assembly and function.
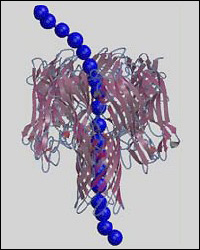
Multiscale model of single-stranded DNA homopolymer through α-hemolysin nanopore.
Because of the multiple time and length scales intrinsically coexisting in biomolecular systems, in the past few decades the field has evolved through parallel lines, focusing at different resolutions. No single technique can at present span the whole range of typical time and length scales relevant for a protein's biochemical function. In order to significantly advance our understanding of the fundamental principles shaping protein mechanisms we are developing new coarse-grained and multi-resolution models and techniques that can overcome the computational impasse. Our line of research has the potential to build computational models to assist molecular design for a variety of applications ranging from drug design to the engineering of novel functional materials.
Professor Matysiak's current protein reserch includes:
- Coarse-grained protein models for the characterization of folding landscapes
- Translocation of polymer through nanopores
- Multiscale modeling techniques
- Cold denaturation
- Characterizing the role of water in biomolecular systems
For more information, please visit Professor Matysiak's web site
- BIOE 331: Biofluids
- BIOE 689D: Computational Molecular Bioengineering
- “Effects of Sequence and Solvation on the Temperature-Pressure Conformational Landscape of Proteinlike Heteropolymers”, Matysiak, S. and Das, P., Phys. Rev. Lett., 111, 058103 (2013) (Online)
- "Thermal Stability of Hydrophobic Helical Oligomers", Romero-Vargas Castrillón, S., Matysiak, S., Stillinger, F., Rossky, P. J., Debenedetti, P., J. Phys. Chem. B, 116, 9963-9970 (2012). (Online)
- "Phase Behavior of a Lattice Hydrophobic Oligomer in Explicit Water", Romero-Vargas Castrillón, S., Matysiak, S., Stillinger, F., Rossky, P. J., Debenedetti, P., J. Phys. Chem. B, 116, 9540-9548 (2012). (Online)
- "The Role of Hydrophobic Hydration in Protein Stability: A 3D Water-Explicit Protein Model Exhibiting Cold and Heat Denaturation", Matysiak, S., Debenedetti, P. G., Rossky, P. J., J. Phys. Chem. B, 116, 8095-8104 (2012). (Online)
- "Direct Characterization of Hydrophobic Hydration during Cold and Pressure Denaturation", Das, P., Matysiak, S., J. Phys. Chem. B, 116, 5342-5348 (2012). (Online)
- "Dissecting the Energetics of Hydrophobic Hydration of Polypeptides", Matysiak, S., Debenedetti, P. G., Rossky, P. J., J. Phys. Chem. B, 115, 14859-65(2011). (Online)
- "Mapping folding energy landscapes with theory and experiment", Matysiak S., Clementi C., Arch. Biochem. Biophys., 469, 29-33 (2008). (Online)
- "Modeling Diffusive Dynamics in Adaptive Simulation of Liquid Water", Matysiak S., Clementi C., Praprotnik M., Delle Site L., Kremer K., J. Chem. Phys. 128,1 (2008). (Online)
- "Adaptive Resolution Simulation of Liquid Water", Praprotnik M., Matysiak S., Delle Site L., Kremer K., Clementi C., J. Phys.: Cond. Matter, 19, 292201 (2007). (Online)
- "Minimalist protein model as a diagnostic tool for misfolding and aggregation", Matysiak S., Clementi C., J. Mol. Biol. 363, 297-308 (2006). (Online)
- "Dynamics of polymer translocation through nanopore: Theory meets experiments", Matysiak S., Montesi A., Kolomeisky A. B., Pasquali M., Phys. Rev. Lett., 96, 118103 (2006). (Online)
- "Balancing energy and entropy: A new minimalist model for the characterization of protein folding landscapes", Das P., Matysiak S., Clementi C., PNAS, 102, 10141-10146 (2005). (Online)
- "Optimal combination of theory and experiments for the characterization of the protein folding landscape of S6", Matysiak S., Clementi C., J. Mol. Biol. 343, 235 (2004). (Online)